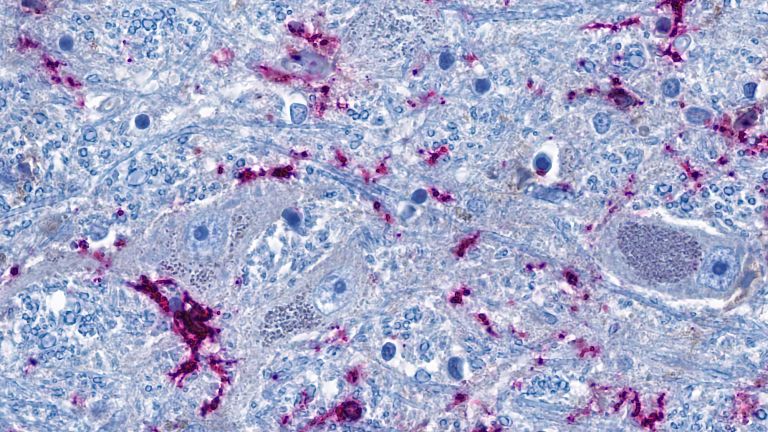
Prionen: Eiweiße, die wahnsinnig machen
Für eine Handvoll besonderer Proteine scheinen die Regeln der Biologie nicht zu gelten. Wie sie den Rinderwahn verursachen und auf Menschen überspringen konnten, ist eine gruselige Geschichte
Wissenschaftliche Betreuung: Dr. Hermann Altmeppen
Veröffentlicht: 02.05.2023
Niveau: leicht
- Prionen sind infektiöse Eiweiße, deren Übertragung auch ohne Beteiligung von Erbsubstanz eine Reihe von Krankheiten auszulösen vermag. Dies geschieht nach Art einer Kettenreaktion, bei der die falsch gefalteten krankmachende Prionen ihr natürliches Gegenstück, das zelluläre Prionprotein, umfalten. So entstehen Aggregate und Nervenzellen sterben ab.
- Die Prionenhypothese, aufgestellt von Stanley Prusiner, war lange umstritten, wurde jedoch im Jahr 1997 mit dem Nobelpreis für Medizin ausgezeichnet und vielfach experimentell untermauert.
- Prionen-Krankheiten bei Tieren sind beispielsweise Scrapie (bei Schafen und Ziegen), die durch Verfütterung von Schafskadavern übertragene Bovine Spongiforme Encephalitis (BSE) bei Rindern oder die „Chronic Wasting Disease“ bei Hirschen, Elchen und Rehen.
- Übertragbare Prionenkrankheiten beim Menschen sind Kuru, das unter bestimmten Urvölkern auf Neuguinea durch rituellen Kannibalismus übertragen wurde, und die „variante Creutzfeld-Jakob-Erkrankung“ (vCJD), die durch verseuchte Rinderprodukte Mitte der 1980er Jahre ausgelöst wurde und mittlerweile über 200 Todesopfer gefordert hat. Auch über ärztliches Handeln, etwa bei der Transplantation der Hornhaut im Auge, wurden Prionenerkrankungen unbeabsichtigt übertragen.
- Beim Menschen treten zudem erbliche Prionen-Erkrankungen auf, wie die familiäre Form der CJD, das Gerstmann-Sträussler-Scheinker (GSS)-Syndrom und die Fatale Familiäre Insomnie (FFI). Ursache sind hier bestimmte Mutationen in dem Gen (PRNP), das für das Prionprotein kodiert. Es handelt sich um autosomal-dominante Erbkrankheiten.
- Die sporadische Form von CJD, die ohne erkennbare genetische oder äußere Ursachen auftritt, macht mit Abstand den größten Anteil aller humanen Prionenerkrankungen aus.
- Bei mehreren neurodegenerativen Krankheiten (Alzheimer, Parkinson, Huntington) bilden sich aus falsch gefalteten Proteinen auch große Aggregate und Ablagerungen im Gehirn. Verschiedene Formen der fehlgefalteten Proteine führen hier ebenfalls zum Untergang von Synapsen und dem Tod von Nervenzellen. Trotz dieser Ähnlichkeiten sind diese Krankheiten aber nicht im herkömmlichen Sinne ansteckend. Eine Zuordnung zu den Prionenerkrankungen ist umstritten.
Wäre Stanley Prusiner einige hundert Jahre früher geboren, hätte man ihn womöglich als Ketzer verbrannt. Oder zumindest aus der Gemeinschaft der Gläubigen ausgestoßen. Denn der Virologe an der Universität Kalifornien in San Francisco und Berkeley hatte an dem Glaubensbekenntnis seiner Zunft gerührt. Das „Zentrale Dogma der Molekularbiologie“ stammt aus dem Jahr 1958 und wurde von keinem geringeren als Francis Crick formuliert, einem Mitbegründer der modernen Biologie und eher unbescheidenen Genie. Dieses Dogma besagt, dass Informationen in biologischen Systemen stets von der Erbsubstanz DNA zum Botenstoff RNA fließen und schließlich in Proteine umgesetzt werden. Die Vorstellung, dass ein Protein genügen könnte, um eine Krankheit zu übertragen, war damit eigentlich unvereinbar. Doch genau dies hatte Prusiner behauptet. Oder zumindest hatte er diese Hypothese weiter verfolgt und ausgebaut. Denn genau genommen, kam die Idee vom infektiösen Protein bereits in den 1960er Jahren auf.
Im Zentrum seines Interesses standen zunächst Schafe, Studienobjekt, die an Scrapie (von engl. „kratzen“) verendet waren. Seit dem 18. Jahrhundert war diese tödliche Tierseuche mit ihren Gang- und Verhaltensstörungen in Europa bekannt, und auch das von Hohlräumen durchlöcherte Gehirn der Tiere war immer wieder beschrieben worden.
Mit Gewebeproben und Blut ließ sich das Leiden im Labor von kranken auf gesunde Schafe übertragen. Auch auf Ziegen, Nerze, Ratten und Mäuse. Ähnliches war US-Forschern bereits 1972 gelungen, als sie infektiöses Material aus dem Gehirn von Schafen, zunächst auf Mäuse und dann vom Mäusegehirn auf Javaneraffen (Macaca fascicularis) übertragen hatten – ein frühes Schlüsselexperiment, auch wenn die Autoren der Studie davon ausgingen, dass ein Virus hinter der Erkrankung steckte.
Dieser Verdacht bestätigte sich jedoch nicht. Der Erreger war ungewöhnlich widerstandsfähig, schlüpfte auch noch durch die engmaschigsten Filter und überdauerte verschiedenste Methoden zur Inaktivierung von Nukleinsäuren. Gab man hingegen eiweißspaltende Enzyme (Proteasen) hinzu, so verloren die Gewebeproben zumindest teilweise ihre Infektiosität. 1982 hatte Prusiner genug Beweise gesammelt. Er veröffentlichte seine Entdeckung in der Fachzeitschrift „Science“ unter der Überschrift: „Novel proteinaceous infectious particles cause scrapie“ – „Neue proteinartige infektiöse Partikel verursachen Scrapie“. Die mysteriösen Erreger nannte Prusiner „Prion“, abgeleitet vom englischen „proteinaceous infectious particles“ (proteinartige infektiöse Partikel).
Nobelpreis für einen Außenseiter
Etwa zehn weitere Jahre verbrachte der wissenschaftliche Außenseiter darauf, seine Prionenhypothese gegen die Zweifel und die Angriffe vieler forschender Kollegen zu verteidigen. Doch er sollte recht behalten und wurde – auch als Anerkennung für sein Durchhaltevermögen – 1997 mit dem Nobelpreis für Physiologie oder Medizin geehrt. Heute weiß man: Der Bauplan von Prion-Proteinen ist zwar genauso in einer Reihenfolge von DNA-Bausteinen festgeschrieben wie bei „normalen“ Proteinen. Der Weg von der DNA über die RNA zum Protein ist also durchaus, dem Dogma entsprechend, gegeben. Allerdings kann das im Genom kodierte Prionprotein (PrP) eine alternative Konformation, also eine alternative dreidimensionale Faltung, annehmen, mit weiteren Prionproteinen verklumpen und seine falsche Struktur dann wiederum normalen Prionproteinen aufzwingen. In seltenen Fällen und oftmals über lange Zeiträume hinweg kann dies zu einer über viele Jahre unbemerkten fatalen Kettenreaktion führen.
All dies wäre womöglich nur eine Randnotiz in der Veterinärmedizin geblieben, hätte man nicht eine ganze Reihe weiterer Krankheiten entdeckt, die Scrapie ähneln und auch Menschen betreffen können. Das Interesse des Humanmediziners Prusiner wurde nach eigenen Angaben geweckt, als ein Patient mit der seltenen Creutzfeldt-Jakob-Erkrankung (CJD) in seiner Obhut verstorben war. Typisch für diese bis heute unheilbare Krankheit sind Bewegungsstörungen, Gedächtnisverlust und Verwirrtheit, sowie ein schneller Verlauf, der in der Regel nach vier Monaten bis zwei Jahren tödlich endet.
Fasziniert war der Neurologe auch von Gerüchten über eine geheimnisvolle Krankheit namens Kuru, die unter einigen Urvölkern in der Ost-Hälfte Neuguineas (heute Papua-Neuguinea) kursierte, und dort bis etwa 1950 offenbar durch rituellen Kannibalismus verbreitet wurde. Die ersten Weißen, die 1933 ins Hochland vordrangen, trafen dort buchstäblich auf eine vergessene Welt. Die Menschen dort lebten noch in der Steinzeit, gehörten Dutzenden von Völkern an, die sich gegenseitig bekriegten und unterschiedliche Sprachen und Dialekte hatten.
Forscher unter Kannibalen
Dass Kannibalismus weit verbreitet war, bestätigen zahlreiche Berichte nicht nur von Missionaren, sondern auch von Anthropologen und der berühmten US-amerikanischen Ethnologin Margaret Mead . Nach einem Besuch des Ortes Timbunke am Fluss Sepik schrieb sie: „Der Fluss steht für Moskitos, Krokodile, Kannibalen und im Wasser treibende Leichen – und ich kann versichern, ich habe alles gesehen.“ Michael Rockefeller , Mitglied der bekannten US-Familie und Hobby-Ethnograf verschwand 1961 an der Südküste Neuguineas und wurde seinem Biographen zufolge von Angehörigen des Volkes des Asmat umgebracht.
Schließlich reiste auch Prusiner nach Neuguinea. Im Jahr 1982 traf er dort auf Carleton Gajdusek , einen Überflieger, der Physik, Chemie, Mathematik studiert, bei den Nobelpreisträgern Linus Pauling und Frank Burnet gelernt und schließlich in Harvard seinen Doktor der Medizin gemacht hatte. Gajdusek war dem Erreger von Kuru auf der Spur und hatte die Krankheit 1966 bereits auf Schimpansen übertragen. Zwei Jahre später gelang ihm das mit CJD, und 1972 mit Scrapie. Ebenso wie Prusiner hat auch Gajdusek den Medizin-Nobelpreis erhalten, allerdings bereits einige Jahre früher, im Jahr 1976. Die beiden Wissenschaftler haben sich laut Prusiners Erinnerungen nicht besonders gut vertragen. Sie waren wohl eher Konkurrenten, denn Kollegen, wie aus einem Rückblick hervorgeht , den Prusiner im Jahr 2008 veröffentlicht hat.
Kuru, eine Krankheit, die nach dem Ausbruch schnell zum Tod führte, hatte offenbar einiges mit CJD und Scrapie gemeinsam, wie Prusiner und andere Kollegen erkannten. Das Zittern und andere Bewegungsstörungen, der bei Kuru eine Demenz folgte, hatten beide Mediziner beobachtet. Für Gajdusek war die Krankheit jedoch der Parkinsonkrankheit ähnlich – für Prusiner nicht. Der wichtigste Streitpunkt aber war, dass Gadjusek nicht an Prusiners Prionen-Hypothese glaubte. Als Prusiner in einer Publikation über Kuru Gajdusek als Co-Autor nennen wollte, erlaubte Gadjusek dies erst, nachdem das Wörtchen „Prionen“ aus dem Artikel getilgt worden war.
Gajduseks machte, jenseits der Wissenschaft, in negativer Weise von sich reden: Von seinen Reisen in die Tropen hatte er – mit Einverständnis der Eltern – insgesamt 56 Kinder mitgebracht und bei sich zu Hause in einer Art Kommune erzogen. Als Vorwürfe von sexuellem Missbrauch laut wurden, bezeugte dies einer seiner „Schützlinge“, der mittlerweile studierte und im Alter von 14 Jahren mit Gadjusek zusammengelebt hatte. Der Forscher wurde daraufhin zu einem Jahr Gefängnis verurteilt, verlor seine Position als Leiter des Hirnlabors am Nationalen Schlaganfallinstitut (NINDS) und lebte nach Verbüßung seiner Strafe bis zum Tod in Paris, Amsterdam und Tromsö, was er nach eigenen Angaben bevorzugte, weil er dort in der ständigen Dunkelheit des Winters besser arbeiten konnte.
Heute werden Scrapie, Kuru und die übertragbare Variante der CJD (es gibt auch eine vererbte und eine spontan auftretende Form) aufgrund ihrer Pathologie als „Transmissible spongiforme Enzephalopathien“ (TSE) eingeordnet – zu Deutsch: „Übertragbare schwammartige Hirnleiden“. Auch der zusammenfassende Begriff „Prionenerkrankungen“ ist geläufig. Die wohl bekannteste aller TSE wurde erstmals 1984 in England nachgewiesen und als „Bovine spongiforme Enzephalopathie“ (BSE) benannt. Besser bekannt ist diese Tierkrankheit unter dem Namen Rinderwahn.
BSE entstammte einer gefährlichen Mixtur von unnatürlichen Praktiken der Fleischerzeugung sowie routinemäßigen Gesetzesverstößen und Vertuschungsversuchen, wie aus Zeugenbefragungen in einem offiziellen Untersuchungsausschuss hervorging. So hatte man Fleisch und Knochenmehl aus Schafskadavern quasi als „Kraftfutter“ an Rinder verfüttert. Bei der eigentlich vorgeschriebenen Sterilisation durch Hitze und Druck wurde geschlampt – oder gar zwecks Kostenoptimierung manipuliert, sodass Scrapie-Erreger aus infizierten Schafen überlebten, vom Magen der Rinder ins Nervensystem gelangten, sich an den neuen Wirt anpassten und die Rinder töteten. Fast 200.000 Tiere verendeten an BSE, und in der gesamten EU wurden fast vier Millionen Rinder gekeult.
Leichtsinn und Skrupellosigkeit am Rande der Apokalypse
Wenn Scrapie von Schafen auf Rinder übertragbar ist, besteht dann nicht auch die Gefahr einer Übertragung von BSE auf den Menschen? Die Befürchtung lag aus heutiger Sicht nahe. Dennoch gelangten verseuchte Hamburger, Würste und andere Fleischprodukte in die Mägen der Verbraucher in aller Welt. Gelatine aus Haut und Knochen wurde zu Pudding, Tortenbelag oder Gummibärchen verarbeitet. Und es dauerte 10 Jahre von den ersten Warnungen der Experten, bis die britische Regierung 1996 einräumte, dass da möglicherweise doch eine Verbindung bestehe zwischen dem Rinderwahn und einer neuen Variante der CJD, an der zu diesem Zeitpunkt bereits 10 Menschen gestorben waren. Nach aktuellem Stand sind es weltweit mehr als 200 Tote, davon die weitaus meisten in Großbritannien, knapp 30 in Frankreich, aber noch kein einziger Fall in Deutschland. Die Menschheit hat offenbar Glück gehabt. Wenn BSE ebenso leicht auf den Menschen übergesprungen wäre, wie viele Viren das tun, hätte es womöglich Millionen von Toten gegeben.

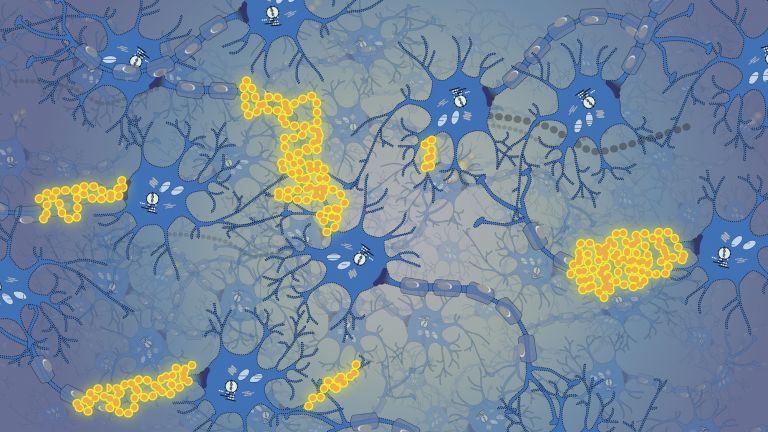
Viele Krankheiten – ein Mechanismus
Die durch Prionen von Rindern auf Menschen übertragene Form der CJD wird als vCJD (für „Variante“) gekennzeichnet. Und es gibt noch eine weitere Form, die iatrogene CJD (iCJD) mit einer eher kleinen Zahl von dokumentierten Fällen, bei denen die Krankheit durch medizinische Prozeduren wie die Verpflanzung von Hirnhäuten, Augenhornhaut oder der Gabe von Wachstumshormonpräparaten von Mensch zu Mensch übertragen wurde.
Alle bisher genannten Prionenkrankheiten verbreiten sich mittels Übertragung. Sie machen aber zusammen weniger als ein Prozent aller Fälle aus. Mit mehr als 85 Prozent ist die sporadische Creutzfeldt-Jakob-Krankheit unter den menschlichen Prionenerkrankungen am häufigsten, und für etwa zwei Todesfälle unter je 1 Millionen Einwohner pro Jahr verantwortlich. Der Rest – rund 15 Prozent – verteilt sich auf erbliche, also genetisch bedingte Prionenerkrankungen wie die familiäre Form der CJD, das Gerstmann-Sträussler-Scheinker (GSS)-Syndrom und die Fatale Familiäre Insomnie (FFI).
Bei den erblichen Prionen-Erkrankungen ist die Reihenfolge der Bausteine in dem natürlichen Gen PRNP auf Chromosom 20 verändert. Inzwischen kennt man mehr als 60 Varianten davon. Ein Vergleich der Genome von über 16.000 Patienten mit rund 600.000 nicht Betroffenen in drei Datenbanken ergab, dass nur wenige Varianten unweigerlich zum Ausbruch der Krankheit führen, viele Varianten dagegen auch bei gesunden Menschen auftraten. Bei den letzteren Fällen müssen offenbar weitere ungünstige Umstände hinzukommen, wie möglicherweise Umweltgifte oder Vorbelastungen durch andere Krankheiten.
Zentral für das Verständnis der Prionenkrankheiten sind die Prionproteine selbst. Die krankhafte, falsch gefaltete und verklumpte Form wird als PrPSc bezeichnet – wobei das „Sc“ auf die Schafskrankheit Scrapie verweist –, und die normale, „gesunde“ Form als zelluläres PrPC . Gelangen einige wenige PrPSc in eine Umgebung voller PrPC so werden auch die „gesunden“ Moleküle umgeformt und die Kettenreaktion nimmt ihren Lauf. Die Konzentration von PrPSC im Hirn und Rückenmark steigt rapide an, und führt schließlich zum Tod von Nervenzellen.
Neuron
Neuron/-/neuron
Das Neuron ist eine Zelle des Körpers, die auf Signalübertragung spezialisiert ist. Sie wird charakterisiert durch den Empfang und die Weiterleitung elektrischer oder chemischer Signale.
Eine verwirrende Vielfalt von Funktionen
Zwar gibt es Dutzende von Forschungsarbeiten zur Funktion des Proteinproteins . Diese sind jedoch oftmals widersprüchlich oder nicht ausreichend, um das Absterben der Nervenzellen zu begreifen – geschweige denn zu verhindern. PrPC zum Beispiel ist, laut einer aktuellen Übersichtsarbeit , bei der neuronalen Entwicklung beteiligt, bei der Zelladhäsion, der Neuroprotektion, der Regulation zirkadianer Rhythmen, der Aufrechterhaltung der Ionenhomöostase usw. Zwar kennt man inzwischen recht genau die Reaktionswege beim Umfaltungsprozess. Und auch die dreidimensionalen Strukturen der beteiligten Eiweißfragmente und Zwischenprodukte sind zunehmend bekannt. Nachgewiesen ist zudem, dass PrPSc bei über die Nahrung erfolgter Übertragung – etwa beim Verzehr von BSE-kontaminiertem Rindfleisch – offenbar vom Magen her in das Nervensystem gelangt und sich dort auf der Membran von Nervenzellen festsetzt. Wie genau es von diesem Punkt aus weitergeht, ist jedoch ziemlich unklar.
Wer nun denkt, dass Stanley Prusiner nach dem Nobelpreis und der Bestätigung seiner Prionenhypothese zufrieden die Hände in den Schoss legt, der irrt. Nach seiner Auffassung könnten Prionen, oder vielmehr ein prionenartiger Mechanismus von sich ausbreitender Eiweißfehlfaltung im Gehirn auch bei anderen neurodegenerativen Erkrankungen am Werk sein. Sowohl bei der Alzheimer-Demenz als auch der Parkinson-Krankheit und bei Chorea Huntington spielen jeweils unterschiedliche Proteine eine Rolle (Aß, Tau, α-Synuklein und Huntingtin), deren Verhalten doch sehr an das von Prionen erinnere. Die Parallelen werden von vielen Forschern durchaus anerkannt. Zwar gibt es keine harten Daten, dass auch nur eine einzige dieser Krankheiten im herkömmlichen Sinne ansteckend sein könnte, doch lauter Widerspruch gegen Stanley Prusiner ist seltener geworden. Im „Harrison“, dem wichtigsten Lehrbuch der Medizin, verantwortet Prusiner das Kapitel über Prionenkrankheiten – eine Art Ritterschlag, die bezeugt, dass dem ehemaligen Außenseiter längst wissenschaftliche Anerkennung zuteilwird.
Zum Weiterlesen
- Sigurdson CJ et al. Cellular and Molecular Mechanisms of Prion Disease. Annu Rev Pathol. 2019 Jan 24;14:497-516. doi: 10.1146/annurev-pathmechdis-012418-013109 (zum Volltext)
- Prusiner SB, Miller BL. Prion Diseases. In: Jameson J, Fauci AS, Kasper DL, Hauser SL, Longo DL, Loscalzo J. eds. Harrison's Principles of Internal Medicine, 20e. McGraw Hill; 2018. (zum Buchkapitel)